Computer-aided drug design
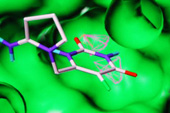
We seek to apply our knowledge of modelling ligand-receptor interactions to adopt a computationally-led design of small molecule modulators. We currently use a range of computational techniques, for example mapping, de novo methods and virtual screening (using molecular docking).
In this way, we pursue a number of multidisciplinary design projects, collaborating with medicinal chemists, biochemists and biologists: one example is our structure-based virtual screening of compound libraries which led to the identification of a novel non-nucleobase-derived inhibitor of human thymidine phosphorylase, an anti-cancer target, as a candidate for lead optimisation.
Selected publications
- Synthesis and biological evaluation of coumarin-based inhibitors of NAD(P)H: Quinone Oxidoreductase-1 (NQO1). K. A. Nolan, J. R. Doncaster, M. Dunstan, K. A. Scott, A. D. Frenkel, D. Siegel, D. Ross, J. Barnes, C. Levy, D. Leys, R. C. Whitehead, I. J. Stratford and R. A. Bryce J. Med. Chem. 2009, 52, 7142-7156.
- Discovery of novel inhibitors of Trypanosoma cruzi trans-sialidase from in silico screening. J. Neres, M. Brewer, L. Ratier, P. N. Edwards, H. Botti, A. Buschiazzo, P. M. Alzari, A. C. C. Frasch, R. A. Bryce and K. T. Douglas. Bioorg. Med. Chem. Lett. 2009, 19, 589-596.